MoIUNet++:自适应粒度显式子结构与互作感知分子表示学习

打开文本图片集
中图分类号:064 doi: 10.1016/j.actphy.2025.100209
MolUNet++: Adaptive-grained explicit substructure interaction aware molecular representation learning
Fing Xu1 , Zhiwei Yang 2*, Sirui wu 3, Wu Su 1, Lizhuo Wang1,Deyu Meng 4,5,* , Jiangang Long 1,* 1 , , 710049, Province, . 2 , , 710049, Province, . 3 ,Ltd, 710o77, Province, . 4 Research Institute for Mathematical , , 710049, Province, . 5 , , , Province, .
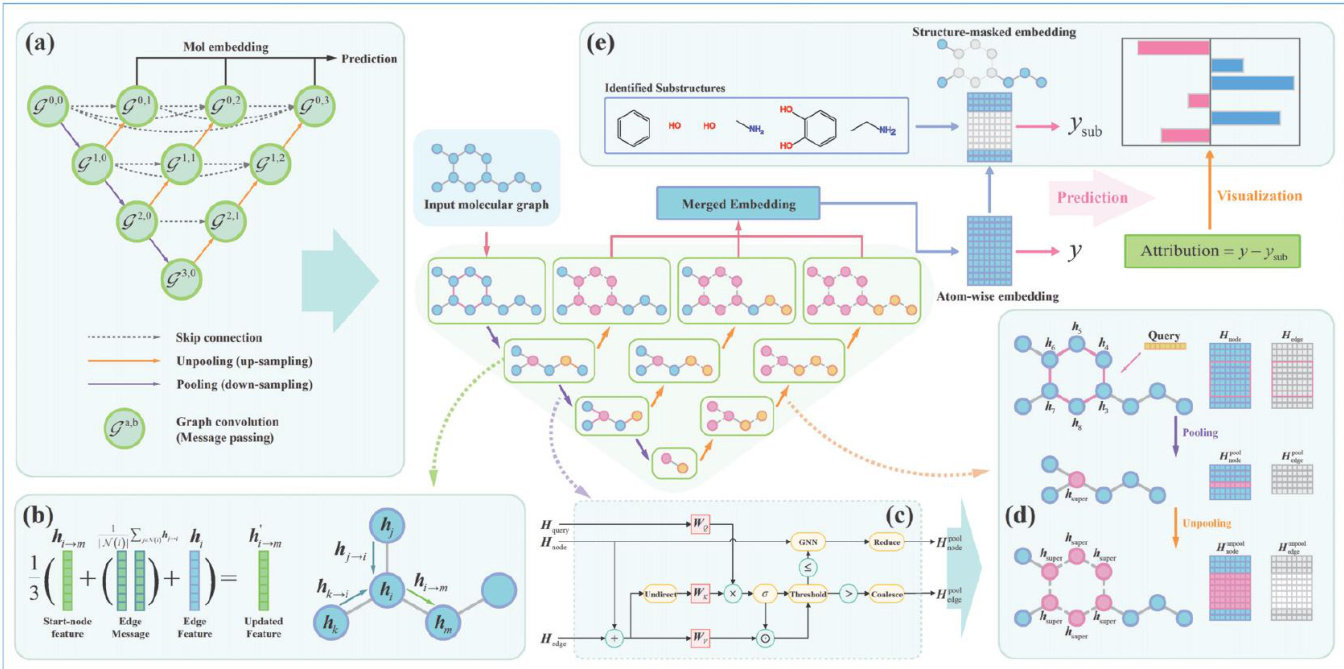
Abstract:Molecular representation learning is a critical task in AI-driven drug development. While graph neural networks (GNNs) have demonstrated strong performance gained widespread adoption in this field, efficiently extracting explicitly analyzing functional groups remains a challenge. To addressthis issue, we propose MolUNet ++ ,a novel model that employs Molecular Edge Shrinkage Pooling (MESPool) for hierarchical substructure extraction, utilizes a Nested UNet framework for multi-granularity feature integration, incorporates a substructure masking explainer for quantitative fragment analysis.We evaluated MolUNet++ on tasks including molecular property prediction, drug-drug interaction (DDI) prediction, drug-target interaction (DTI) prediction. Experimental results demonstrate that MolUNet ++ not only outperforms traditional GNN models in predictive performance but also exhibits explicit, intuitive, chemically logical interpretability.This capability provides valuable insights tools for researchers in drug design optimization.
Key Words:Molecular representation learning;GNN; Structure identification; Adaptive granularity
1引言
分子表示在连接生物信息学、化学信息学、材料科学以及尤其是药物发现等多个科学领域与机器学习之间,起着至关重要的桥梁作用[1-3]。(剩余26709字)